Claude Science Review: Bioinformatics Paper Reproduction — What Reproduced and What Didn't
Claude Science reproduced Figures 1–14 of a breast cancer bioinformatics paper using Opus alone — 2 skills, 0 MCP calls. Here's what worked, what didn't, and why it matters.
TL;DR
- Only 2 skills invoked (pdf-explore and figure-style), MCP calls: 0 — the entire 14-figure analysis ran on Opus model capability alone. The model handled it impressively, but at significant token cost.
- Claude Science reproduced the full in-silico backbone of a breast cancer bioinformatics paper (Figures 1–14) using an independent R pipeline
- Key claims matched exactly: mutation rate (9.69%), PTEN as top mutant, AMPK as top KEGG pathway, four-gene survival model direction
- Key divergences: DEG count (20,156 vs. paper's 3,492), consensus clustering split (720/398 vs. 865/253), LASSO gene selection (3 genes vs. 4)
- The right metric for scientific agents is auditability, not polish
I tested Claude Science on a full bioinformatics paper reproduction task. The target was Xu et al. 2025 in the Journal of Translational Medicine, a breast cancer study that proposed drug-resistance and mitochondrial-energy-metabolism-related differentially expressed genes as prognostic markers.
This is a better stress test for scientific agents than a simple Q&A benchmark. The task was not just "read a paper and summarize it." It required following a long, dependent computational workflow: data retrieval, preprocessing, differential expression analysis, mutation and CNV analysis, enrichment, consensus clustering, immune microenvironment analysis, drug sensitivity prediction, PPI network construction, LASSO-Cox modeling, survival validation, nomogram construction, and comparison against the published figures. The result was encouraging, but also a useful reminder of what scientific reproduction actually means.
What the Original Paper Tried to Do
The paper studied breast cancer through the intersection of two biological themes: drug resistance and mitochondrial energy metabolism.
The authors collected drug-resistance-related genes from DRESIS and MSigDB, mitochondrial-energy-metabolism-related genes from GeneCards and prior literature, and differentially expressed genes from TCGA-BRCA and GEO datasets. By intersecting these sources, they identified 15 DMRDEGs: drug-resistance and mitochondrial-energy-metabolism-related differentially expressed genes.
They then used these genes to analyze mutation and CNV patterns, functional enrichment, molecular subtypes, immune infiltration, immunotherapy response indicators, drug sensitivity, protein interactions, and prognosis. The final computational model centered on a four-gene risk score using ATP7B, FUS, AIFM1, and PPARG. The paper also included wet-lab validation of AIFM1 in breast cancer cell lines.
What Claude Science Reproduced
Claude Science reproduced the complete in-silico backbone of the paper, covering Figures 1–14, using an independent R pipeline and raw public data from TCGA and GEO. The wet-lab AIFM1 knockdown experiments were correctly treated as out of scope.
What Claude Science Reproduced Well
Claude Science matched the paper's core computational claims across four key areas: dataset structure, mutation landscape, pathway enrichment, and survival modeling.
Dataset Structure
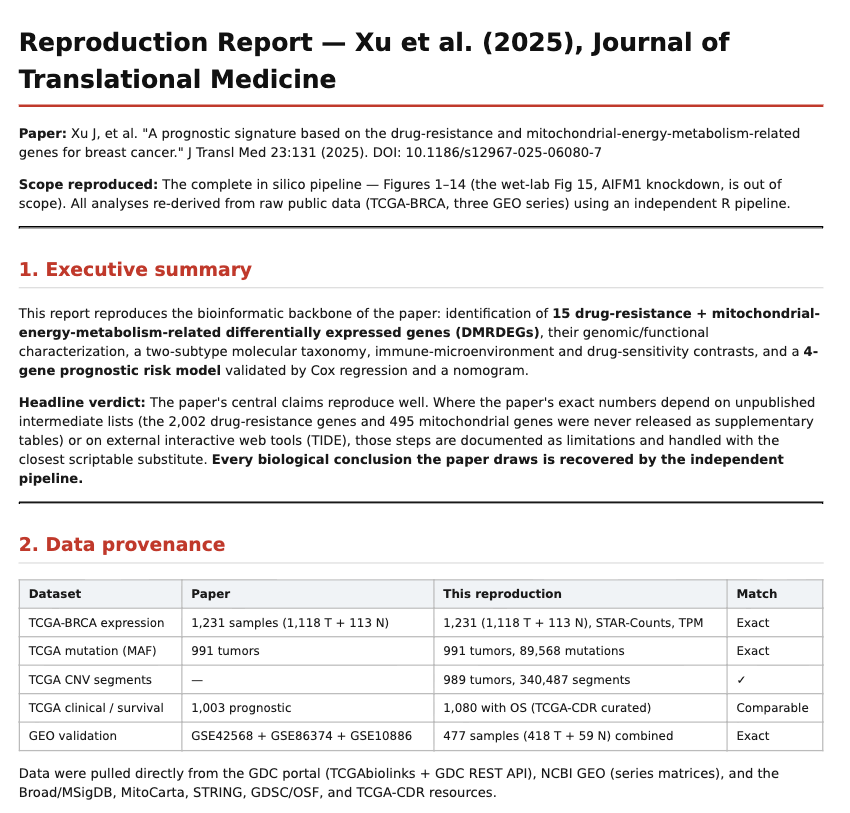
The TCGA-BRCA expression dataset matched the paper’s sample structure:1,231 samples, including 1,118 tumor and 113 normal samples. The GEO validation cohorts also matched the paper’s three-series design.
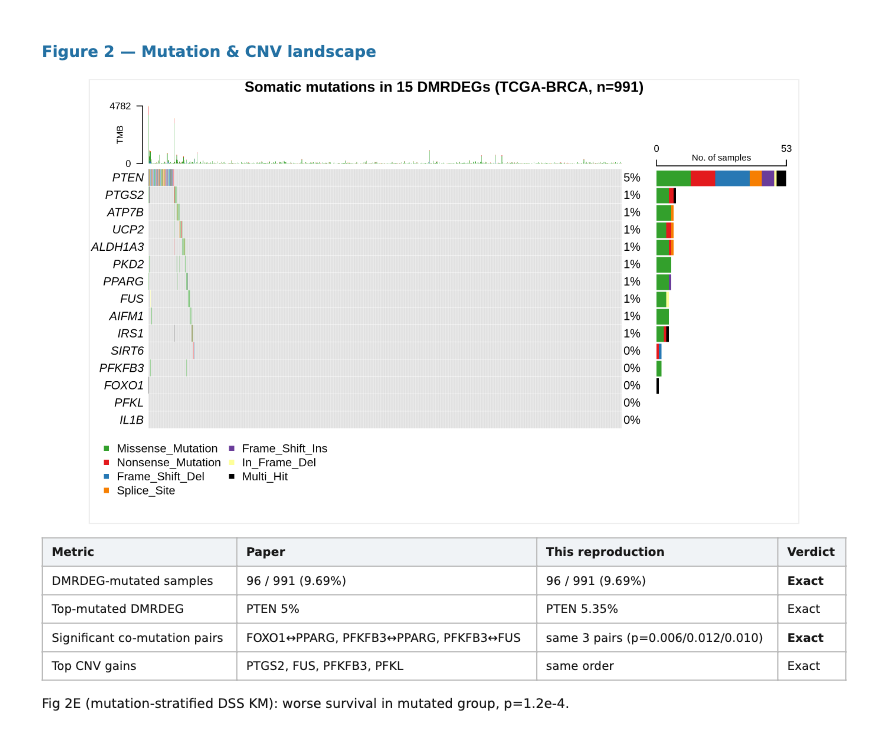
Mutation Landscape
The mutation landscape reproduced almost exactly.The reproduction found 96 out of 991 samples with mutations in the DMRDEG set, matching the paper’s 9.69 percent mutation rate. PTEN was again the top-mutated gene, and the same three significant co-mutation pairs were recovered.
Pathway Enrichment
The functional enrichment result also aligned with the paper's main biological claim. AMPK signaling was recovered as the top KEGG pathway, along with other relevant pathways such as FoxO signaling and central carbon metabolism in cancer.
Survival Modeling
The survival model also reproduced the core conclusion. Using the paper’s published ATP7B/FUS/AIFM1/PPARG formula, the high-risk group had worse overall survival, and the risk score remained independently prognostic in multivariate Cox modeling.

So at the level of the main computational storyline, Claude Science did a strong job. It did not merely generate plausible-looking plots. It executed a long, dependent analysis workflow and recovered many of the paper's central in-silico claims.
Where Exact Reproduction Became Difficult
Four areas could not be exactly reproduced: the upstream gene list intersection, the DEG count, two downstream clustering steps, and two analysis tools that required method substitution. In each case, the divergence traced back to undisclosed implementation details in the original paper, not errors in the reproduction pipeline.
The upstream gene lists were not fully reproducible
The exact upstream gene intersection cannot be reconstructed from public artifacts alone — nosupplementary files were released for the intermediate lists.
The paper reports 2,002 drug-resistance genes and 495 mitochondrial-energy-metabolism-related genes, but the exact intermediate gene lists were not released as supplementary files. GeneCards and DRESIS are also not straightforward to access programmatically. This means that the exact upstream intersection cannot be fully reconstructed from public artifacts alone.
Claude Science handled this by adopting the paper’s 15 DMRDEGs as the canonical downstream set, then checking whether all 15 were genuine, direction-consistent DEGs in the independent DESeq2 run. They were. This was a reasonable and transparent choice, but it should be called an approximate reproduction, not a perfect reconstruction of the original gene-selection process.
The stated DEG threshold did not reproduce the paper's reported DEG count
The paper states a threshold of |log2FC| > 0.5 and adjusted p < 0.05. Applying those thresholds in DESeq2, the reproduction found about 20,156 DEGs, while the paper reported 3,492. The reported count is much closer to what one would expect from a stricter effective fold-change filter.
This is a common problem in bioinformatics reproduction: a paper may describe the broad method, but omit implementation details that strongly affect the result. Filtering order, normalization choices, annotation versions, low-count filtering, batch handling, and hidden thresholds can all change the final gene set.
Not every downstream panel was identical
Consensus clustering did recover a two-subtype structure, but the subtype split differed from the paper. The reproduction produced a 720/398 split, while the paper reported 865/253. This likely reflects sensitivity to preprocessing or gene-centering choices that were not fully specified.
The LASSO-Cox step also differed. The independent run selected three genes rather than the paper’s four-gene model. Because the paper published the final ATP7B/FUS/AIFM1/PPARG risk formula, Claude Science used that published formula for downstream validation. This preserved the intended evaluation of the paper’s model, but it again means the run should be described as a validated approximate reproduction rather than a fully identical rerun.
Some tools required practical substitution
The paper used TIDE for immunotherapy-response analysis, but TIDE is an interactive web tool without a straightforward scriptable API. Claude Science therefore used IPS and TMB to address the same immunotherapy-response question in a reproducible pipeline.
The paper also used pRRophetic for drug sensitivity prediction. The reproduction used oncoPredict/GDSC2, a practical successor for similar drug-sensitivity workflows. This is a reasonable engineering workaround, but scientifically it should be explicitly labeled as a method substitution.
Reproduction Results: What Matched and What Diverged
| Analysis Step | Paper Claim | Reproduction Result | Match | Notes |
|---|---|---|---|---|
| TCGA-BRCA sample count | 1,231 samples (1,118 T + 113 N) | 1,231 (1,118 T + 113 N), STAR-Counts, TPM | ✅ Exact | |
| GEO validation | GSE42568 + GSE86374 + GSE10886 | 477 samples (418 T + 59 N) combined | ✅ Exact | |
| DMRDEG-mutated samples | (96/991) 9.69% | (96/991) 9.69% | ✅ Exact | |
| Top-mutated DMRDEG | PTEN 5% | PTEN 5.35% | ✅ Exact | |
| Co-mutation pairs | 3 significant pairs | Same 3 pairs | ✅ Exact | |
| Top CNV gains | PTGS2, FUS, PFKFB3, PFKL | same order | ✅ Exact | |
| Survival model direction | High-risk = worse OS | High-risk = worse OS | ✅ Directional | Published formula applied |
| Cox multivariate significance | Risk score independent | Risk score independent | ✅ Directional | |
| DEG count | 3,492 (|log2FC| > 0.5, adj.p < 0.05) | ~20,156 | ❌ Diverged | Hidden stricter threshold likely |
| Consensus clustering split | 865 / 253 | 720 / 398 | ⚠️ Partial | Preprocessing sensitivity |
| LASSO-Cox gene selection | 4 genes (ATP7B, FUS, AIFM1, PPARG) | 3 genes | ⚠️ Partial | Published formula used downstream |
| Upstream gene list reconstruction | 2,002 DR genes / 495 MEM genes | Not fully reconstructable | ❌ Diverged | No supplementary files released |
| Immunotherapy analysis | TIDE | IPS + TMB | 🔄 Substituted | TIDE has no scriptable API |
| Drug sensitivity | pRRophetic | oncoPredict / GDSC2 | 🔄 Substituted | Practical successor tool |
What This Says About Scientific AI Agents
My main takeaway is that Claude Science performed well not because it made the final outputs look polished, but because it preserved the difference between what was reproduced, what was approximated, and what could not be exactly reconstructed from the public record. That distinction matters.
For scientific agents, the goal should not be to create a convincing-looking reproduction report at all costs. The goal should be to maintain provenance and claim boundaries across a long workflow. A useful scientific agent should track where the data came from, which version of the dataset was used, which intermediate files were unavailable, which thresholds failed to match the paper, which substitutions were made, which random seeds were fixed, which covariates differed, and which conclusions are supported only directionally rather than exactly. This is where bioinformatics paper reproduction becomes a strong benchmark for agents. The challenge is not just reasoning. It is execution discipline.
A scientific agent has to retrieve data, run code, debug environments, handle missing resources, make conservative methodological decisions, and avoid overstating the result. That is much closer to real scientific work than answering isolated questions about a paper.
There is another important lesson from this run. Claude Science’s result was mostly driven by the base model and the platform’s internal workflow design, not by a large external tool ecosystem. The agent did not heavily rely on specialized domain skills. Only pdf-explore and figure-style were called. MCP usage was zero. That makes the result more interesting in one way and more limited in another. It is interesting because it shows how far a strong model like Opus can go when it is placed inside a structured scientific-workflow environment. It can read the paper, plan analyses, debug code, inspect outputs, revise tasks, and maintain a reasonably conservative interpretation of the result.
But it is also a limitation because mature scientific agents should eventually rely less on raw model capability and more on reusable, validated domain workflows. For medical research and bioinformatics, I would expect future systems to call dedicated skills for DEG analysis, survival modeling, enrichment, immune infiltration, drug sensitivity, provenance checks, and figure-level validation.
The built-in reviewer and notebook were useful pieces of the Claude Science framework. The reviewer helped inspect task quality, while the notebook provided a structured workspace for files and outputs. But these are still scaffolding layers. They are not the same as reusable bioinformatics procedures with explicit assumptions, validation checks, and versioned execution paths.
Conclusion
I would not call this a perfect replication of the Xu et al. 2025 paper. The upstream gene-source reconstruction, DEG count, subtype proportions, LASSO feature selection, and some immunotherapy or drug-sensitivity tools were not identical to the original study.
But I would call it a strong approximate reproduction of the computational backbone. Claude Science recovered the main in-silico structure of the study, reproduced several central claims exactly or directionally, and documented the parts that could not be fully reproduced from the available public information. That is the right behavior for a scientific agent.
The remaining bottleneck is not only model capability. It is also the reproducibility gap in scientific publishing itself. Papers often omit the exact intermediate gene lists, filtering logic, software versions, preprocessing decisions, and tool settings needed for exact reruns. Scientific agents should not only be judged by whether they can produce a final answer or a polished figure. They should be judged by whether their workflow is auditable.
Claude Science did well on that axis. The next step is better workflow memory, provenance tracking, reusable domain skills, and verification loops that make scientific reproduction more transparent rather than merely more automated.
Related reading:
FAQ
Can Claude Science fully reproduce a bioinformatics paper from scratch?
Not fully, but substantially. In this test, Claude Science reproduced the complete in-silico backbone of Xu et al. 2025 — covering 14 figures — using raw public data from TCGA-BRCA and GEO. Several central claims matched exactly (9.69% mutation rate, PTEN as top mutant, AMPK as top KEGG pathway). However, 3 of 14 analysis steps diverged or required tool substitution, primarily due to undisclosed implementation details in the original paper.
What should AI agents do when a paper's methods can't be exactly reproduced?
The right behavior is to document the gap explicitly, adopt a conservative fallback (e.g., using the paper's published downstream gene list rather than reconstructing the intermediate step), and label the result as an approximate reproduction. Claude Science did this correctly. A scientific agent that silently substitutes methods and reports success is more dangerous than one that flags the discrepancy.
Is auditability more important than accuracy for scientific agents?
An agent that tracks every substitution, version, threshold decision, and scope limitation gives researchers something they can interrogate. One that produces polished figures without audit trails cannot be trusted, even if the figures look right.